
Ацил-КоА-дегидрогеназы, также ACADs (сокр. от англ. Acyl-CoA dehydrogenases, КФ 1.3.99.3) — семейство ферментов из класса оксидоредуктаз, которые катализируют реакции переноса протона (дегидрогенизация) от субстрата — ацил-КоА жирной кислоты на электрон-переносящий флавопротеин (FAD), участвуют в процессе β-окисления. Результатом реакции является образование двойной связи расположенной между атомами С2 (α) и С3 (β) в молекуле тиоэфира субстрата (ацил-КоА)[1].
Флавопротеин в данном случае молекула FAD является простетической группой.
Ферменты катализируют реакции β-окисления жирных кислот, протекающие по следующей схеме:

или Ацил-КоА + FAD → транс-2,3-дегидроацил-КоА + FADH2
Образовавшийся продукт реакции тиоэфир ненасыщенной жирной кислоты (транс-Δ2-еноил-КоА) имеет двойную связь в транс-положении.
ACADs могут быть разделены на три группы в зависимости от их специфики для коротко-, средне- и длинноцепочечных ацил-КоА жирных кислот. Несмотря на различия длины цепей субстрата, все виды ACADs механистически подобны. Различия в ферментах происходит на основе расположения активного центра в аминокислотной последовательности[2].
Ферменты ACADs идентифицированы у многих животных (9 важнейших ферментов), а том числе нематод[3], а также у растений[4], грибов[5] и бактерий[6]. Пять из этих девяти ферментов участвуют в β-окислении жирных кислот (SCAD, MCAD, LCAD, VLCAD, и VLCAD2), а остальные четыре участвуют в метаболизме аминокислот с разветвлённой цепью (i3VD, i2VD, GD и iBD). Большинство ацил-КоА-дегидрогеназ являются α4-гомотетрамерами, а в двух случаях (для очень длинноцепочечных жирных кислотных субстратов) они являются α2-гомодимерами. Был обнаружен дополнительный класс ацил-КoA-дегидрогеназ, который катализирует реакции α,β-ненасыщенности с стероил-КoA-тиоэфирами в некоторых типах бактерий[7][8]. Было продемонстрировано, что этот класс ACAD образует гетеротетрамеры α2β2, а не обычный гомотетрамер α4, белковая архитектура, которых развилась для того, чтобы разместить гораздо больший по размеру стероил-КoA-субстрат[9][10].
Структура

Наиболее изученной структурой среди ферментов данной группы является структура Ацил-КоА-дегидрогеназы жирных кислот со средней цепью (MCAD, КФ 1.3.8.7). Она представлена в виде тетрамера, в каждой субъединице которой содержится по 400 аминокислотных остатков и 1 молекула FAD на один мономер. Тетрамер классифицируют как «димер димера», имеющий общий диаметр в 90 Å (9 нм).
Простетическая группа — FAD связывается с тремя доменами мономера, где вносит существенный вклад в общую стабильность фермента. Ацил-КоА жирных кислот полностью связывается с каждым мономером фермента. Активный центр выровнен аминокислотными остатками F252, T255, V259, T96, T99, A100, L103, Y375, Y375 и E376.
MCAD может связываться с довольно широким спектром длины цепей субстратов — ацил-КоА жирных кислот, однако исследования показывают, что наиболее специфичной целью для связывания является октаноил-КоА (С8-КоА)[11].
Механизм катализа
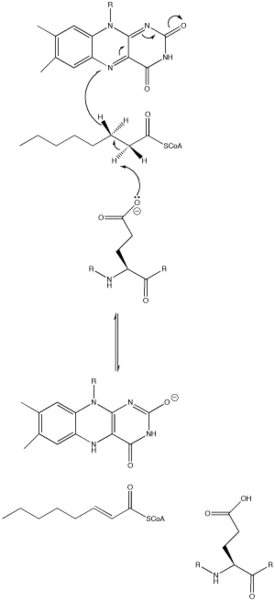
Механизм катализа основывается на реакциях элиминирования E2 (отщепления) двух протонов от субстрата и, последующим их переносом на FAD. Отщепление протонов инициируется остатком глутамата, который, хоть и необходим для механизма протекания реакции, не сохраняется[1].
Остаток глутамата может появляться в самых различных местах в различных видах ацил-КоА-дегидрогеназ (например, для MCAD это Glu-376). Он депротонирует (отщепляет) про-R водород у атома углерода в положении α (C2) в молекуле ацил-КоА. Водородные связи карбонильного кислорода субстрата для обоих 2'-ОН групп рибитола в боковой цепи FAD и основной цепи N—H из ранее упомянутого остатка глутамата снижают рKа (константу кислотности) этого протона, что позволяет ему быстро быть удалённым с субстрата при помощи остатка Glu-376[1].
По мере того как происходит депротонирование альфа-углерода (С2), про-R водород бета-углерода (С3) покидает молекулу субстрата и тот, уже как гидрид анион движется к FAD в согласованной стадии. Протон присоединяется к Re стороне FAD в положении N-5, при этом фермент удерживает FAD на месте посредством водородных связей с участком пиримидина и гидрофобных взаимодействий с диметилбензоловой частью. Субстрат теперь превращается в a,β-ненасыщенные тиоэфир[1].
Как только FAD принимает гидрид анион, атом карбонильного кислорода, прилегающий к атому азота в положении N-1 становится отрицательно заряженным. Эти электроны находятся в резонансе с N-1 атомом азота, которые распространяют и стабилизируют в результирующий отрицательный заряд. Заряд также стабилизируется водородной связью между атомами кислорода и азота и различными аминокислотными остатками активного центра фермента[1].

Дефицитные состояния, связанные с метаболическими нарушениями человека
Дефицитные состояния ацил-КоА-дегидрогеназ приводят к замедлению процессов β-окисления жирных кислот, тем самым показывая, метаболические нарушения. Наиболее частыми являются генетические нарушения такими, как дефицит ацил-КоА-дегидрогеназ жирных кислот со средней длиной цепи (MCADD, сокр. от англ. Medium-chain acyl-CoA dehydrogenase deficiencies) аутосомно-рецессивное заболевание, приводящее к летальным состояниям организма. Некоторые симптомы, характеризующие MCADD: приступы рвоты, гипогликемия и синдром внезапной младенческой смерти (развивается на фоне обильной утилизации глюкозы) и другие. Все эти симптомы непосредственно связаны с накоплением жирных кислот средней цепи (особенно каприловой) и их производных в крови и вторичным дефицитом карнитина. Это приводит к закислению и понижению pH крови и, как следствие, к ацидозу[1][12]. Большую опасность эти проявления MCADD представляют у новорождённых детей, среди них наблюдается самая высокая летальность (до 60 %)[12].
Причиной МСАDD является мутация гена ACADM. Примерно в 90 % случаев она проявляется в замене лизина в позиции 304 (Lys-304) на глутамат, тем самым лишает фермент нормального функционирования. Мутация выявляется у 1 из 20000 новорождённых каждый год. Так как MCADD относится к рецессивным мутациям, то часто родители детей, которые страдают от дефицита могут быть потом диагностированы как носители[13].
Молекулярные основы мутации
В организме человека наиболее распространённые естественные мутации в MCAD происходят по аминокислотному остатку лизина в позиции 304 (Lys-304). В результате точечных мутаций в боковой цепи происходит замена лизина на остаток глутамата. Lys-304, как правило, взаимодействует с окружающими аминокислотными остатками, образуя водородные связи с остатками Gln-342, Asp-300 и Asp-346. Когда происходит мутация и место лизина занимает глутамат, то он вызывает негативное проявление — дополнительный отрицательный заряд (вследствие наличия у последнего двух карбоксильных групп) внедряется на той стороне, где образуется водородная связь, разрушая её. Такое нарушение изменяет складчатую структуру фермента, в конечном счёте, ставя под угрозу его стабильность и подавляя его главную функцию — окисление жирных кислот. Эффективность мутировавшего белка примерно в 10 раз ниже, чем у нативного белка. Это приводит к симптомам MCADD[14].
См. также
Примечания
- ↑ 1 2 3 4 5 6 Thorpe C., Kim J. J. Structure and mechanism of action of the acyl-CoA dehydrogenases (англ.) // The FASEB Journal : journal. — Federation of American Societies for Experimental Biology, 1995. — June (vol. 9, no. 9). — P. 718—725. — PMID 7601336.
- ↑ Kim J. J., Wang M., Paschke R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 1993. — August (vol. 90, no. 16). — P. 7523—7527. — doi:10.1073/pnas.90.16.7523. — PMID 8356049. — PMC 47174.
- ↑ Komuniecki R., Fekete S., Thissen-Parra J. Purification and characterization of the 2‐methyl branched‐chain Acyl-CoA dehydrogenase, an enzyme involved in NADH-dependent enoyl-CoA reduction in anaerobic mitochondria of the nematode, Ascaris suum (англ.) // J Biol Chem : journal. — 1985. — Vol. 260. — P. 4770—4777. — PMID 3988734.
- ↑ Bode, K.; Hooks, M.A.; Couee, I. Identification, separation, and characterization of acyl-coenzyme A dehydrogenases involved in mitochondrial β-oxidation in higher plants (англ.) // Plant Physiology : journal. — American Society of Plant Biologists, 1999. — Vol. 119. — P. 1305—1314. — doi:10.1104/pp.119.4.1305.
- ↑ Kionka, C.; Kunau, W.H. Inducible β-oxidation pathway in Neurospora crassa (англ.) // Journal of Bacteriology : journal. — 1985. — Vol. 161. — P. 153—157.
- ↑ Campbell, J.W.; Cronan, J.E. Jr. The enigmatic Escherichia coli fadE gene is yafH (англ.) // Journal of Bacteriology : journal. — 2002. — Vol. 184, no. 13. — P. 3759—3764. — doi:10.1128/JB.184.13.3759-3764.2002.
- ↑ Thomas, S.T.; Sampson, N.S. Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain (англ.) // Biochemistry : journal. — 2013. — Vol. 52, no. 17. — P. 2895—2904. — doi:10.1021/bi4002979. — PMID 23560677. — PMC 3726044.
- ↑ Wipperman, M.F.; Yang, M.; Thomas, S.T.; Sampson, N.S. Shrinking the FadE Proteome of Mycobacterium tuberculosis: Insights into Cholesterol Metabolism through Identification of an α2β2 Heterotetrameric Acyl Coenzyme A Dehydrogenase Family (англ.) // Journal of Bacteriology : journal. — 2013. — Vol. 195, no. 19. — P. 4331—4341. — doi:10.1128/JB.00502-13. — PMID 23836861. — PMC 3807453.
- ↑ Voskuil, M.I. Mycobacterium tuberculosis Cholesterol Catabolism Requires a New Class of Acyl Coenzyme A Dehydrogenase (англ.) // Journal of Bacteriology : journal. — 2013. — Vol. 195, no. 19. — P. 4319—4321. — doi:10.1128/JB.00867-13. — PMID 23893117. — PMC 3807469.
- ↑ Wipperman, Matthew, F.; Thomas, Suzanne, T.; Sampson, Nicole, S. Pathogen roid rage: Cholesterol utilization by Mycobacterium tuberculosis (англ.) // Crit. Rev. Biochem. Mol. Biol. : journal. — 2014. — Vol. 49, no. 4. — P. 269—293. — doi:10.3109/10409238.2014.895700. — PMID 24611808. Архивировано 28 мая 2020 года.
- ↑ Kieweg V., Kräutle F. G., Nandy A. et al. Biochemical characterization of purified, human recombinant Lys304→Glu medium-chain acyl-CoA dehydrogenase containing the common disease-causing mutation and comparison with the normal enzyme (англ.) // Eur. J. Biochem. : journal. — 1997. — June (vol. 246, no. 2). — P. 548—556. — doi:10.1111/j.1432-1033.1997.00548.x. — PMID 9208949. Архивировано 5 января 2013 года.
- ↑ 1 2 Нельсон Д., Кокс М. Основы биохимии Ленинджера. — М.: БИНОМ, 2011. — Т. II.
- ↑ Touma E. H., Charpentier C. Medium chain acyl-CoA dehydrogenase deficiency (англ.) // Arch. Dis. Child. : journal. — 1992. — January (vol. 67, no. 1). — P. 142—145. — doi:10.1136/adc.67.1.142. — PMID 1739332. — PMC 1793557.
- ↑ Nasser I., Mohsen A. W., Jelesarov I., Vockley J., Macheroux P., Ghisla S. Thermal unfolding of medium-chain acyl-CoA dehydrogenase and iso(3)valeryl-CoA dehydrogenase: study of the effect of genetic defects on enzyme stability (англ.) // Biochim. Biophys. Acta : journal. — 2004. — September (vol. 1690, no. 1). — P. 22—32. — doi:10.1016/j.bbadis.2004.04.008. — PMID 15337167. Архивировано 24 июня 2018 года.
Обычно почти сразу, изредка в течении часа.